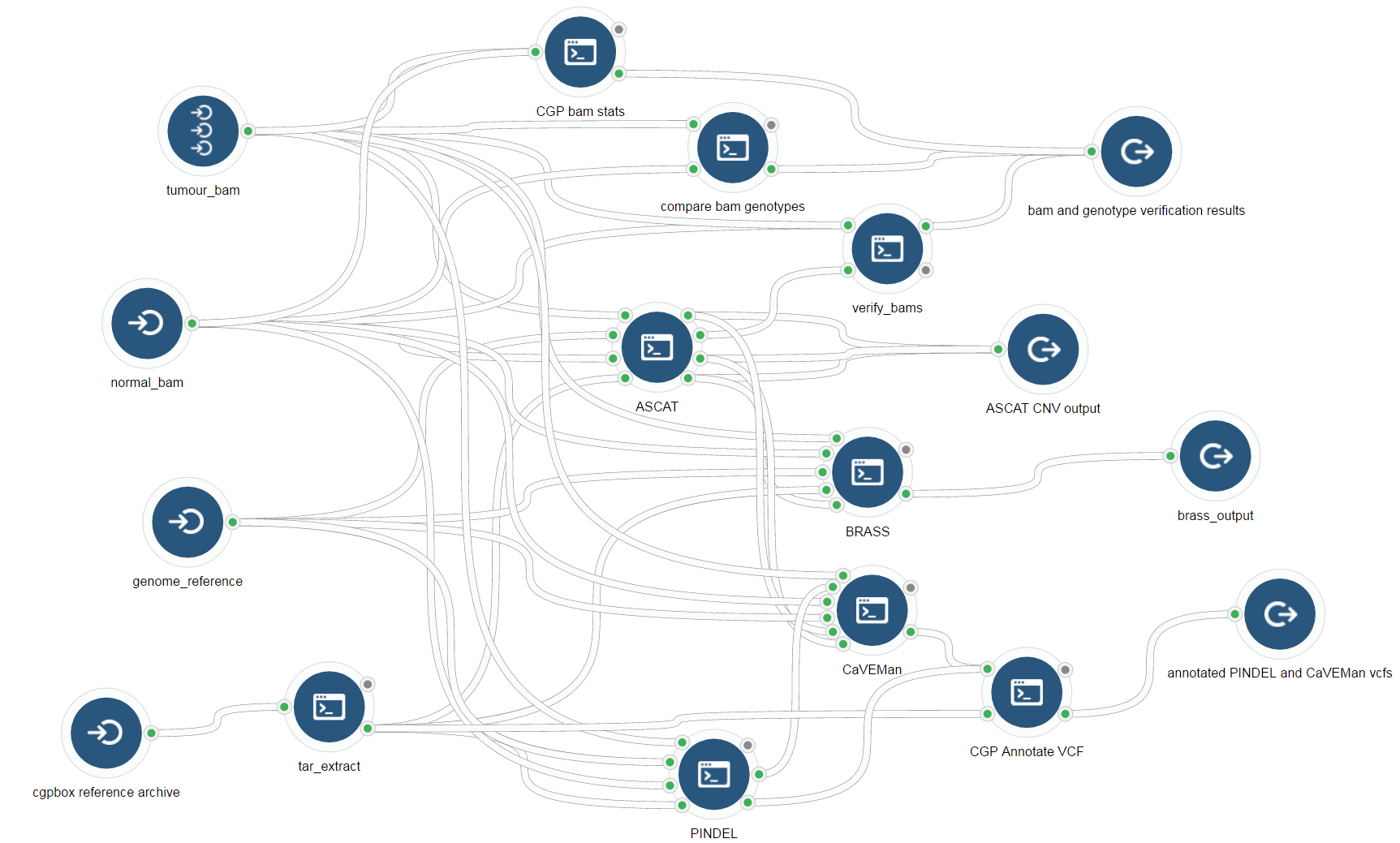
CGPBox Pipeline
Workflow Graph
https://github.com/cancerit/cgpbox
This is a repackaging of the cgpbox pipeline to allow for parellization via the rabix executor and descriptions of the tools included by commmon workflow language.
Descriptions of Individual Tools
1. CGP Bam Stats
Generates useful stats on the wildtype and mutant tumour bams that will be used in the pipeline.
base command
bam_stats -i $BAM_MT_TMP -o $BAM_MT_TMP.basdocker
The docker conatiner is
cgrlab/cgpbox_dev:develop
App ports
Inputs
inputs = list(
input(id = "bam", label = "bam", description = "either wildtype or tumour bam", type = "File", secondaryFiles = list(".bai"), prefix = "-i")
)arguments
arguments = (CCBList(
CommandLineBinding(position = 2, prefix = "-o", valueFrom = list("{return $job.inputs.bam.name + \".bas\"}")),
CommandLineBinding(position = 3, valueFrom = list("\"&& ls -lR \""))
))Outputs
outputs = list(
output(id = "std_out", label = "std_out",
description = "standard output", type = "File...",
metadata = list(org = "cgp"),
glob ="\"std.out\""),
output(id = "bam_statistics", label = "bam stat output",
description = "bam stat output", type = "File",
inheritMetadataFrom = "#bam", metadata = list(org = "cgp"),
glob = Expression(engine = "#cwl-js-engine",
script = "\"*.bas\""))
)Define Tool Object and Push to Platform
Tool object
tool <- Tool(
id = "bam-stats",
label = "CGP bam stats",
hints = requirements(docker(pull = "cgrlab/cgpbox_dev:develop"), cpu(1), mem(1000)),
baseCommand = "bam_stats",
stdout = "std.out",
inputs = inputs,
arguments = arguments,
outputs = outputs)Push app to sbg platform
project$app_add("bam-stats", tool)2. CGP Genotype Check
base command
compareBamGenotypes.pl \
-o /datastore/output/$NAME_WT/genotyped \
-nb $BAM_WT_TMP \
-j /datastore/output/$NAME_WT/genotyped/result.json \
-tb $BAM_MT_TMP \docker
The docker conatiner is
cgrlab/cgpbox_dev:develop
App ports
Inputs
inputs = list(
input(id = "normal_bam", label = "normal_bam", description = "normal wildtype bam", type = "File", prefix = "-nb", stageInput = "link", secondaryFiles = list(".bai")),
input(id = "tumour_bam", label = "tumour_bam", description = "mutant tumour bam", type = "File", prefix = "-tb", stageInput = "link", secondaryFiles = list(".bai"))
)arguments
arguments = CCBList(CommandLineBinding(position = 99, valueFrom = list("\"&& ls -lR\"")))Outputs
outputs = list(
output(id = "std_out", label = "std_out",
description = "standard output", type = "File...",
metadata = list(org = "cgp"),
glob = "\"std.out\""),
output(id = "genotype_comparison_results", label = "genotype comparison results",
description = "genotype comarison results", type = "File",
inheritMetadataFrom = "#tumour_bam", metadata = list(org = "cgp"),
glob = Expression(engine = "#cwl-js-engine",
script = "\"genotype_check/*.json\""))
)Define Tool Object and Push to Platform
Tool object
tool <- Tool(
id = "genotype-check",
label = "compare bam genotypes",
hints = requirements(docker(pull = "cgrlab/cgpbox_dev:develop"), cpu(1), mem(1000)),
baseCommand = "compareBamGenotypes.pl -o genotype_check -j genotype_check/genotype_check_result.json",
stdout = "std.out",
inputs = inputs,
argument = arguments,
outputs = outputs)Make cwl file
write(tool$toJSON(pretty = TRUE), "genotype_check.json")Push app to sbg platform
project$app_add("genotype-check", tool)3. Verify Bams
base command
verifyBamHomChk.pl
-b [bam file]
-a [tumour only - from ASCAT tool - called copynumber.caveman.csv ]
-d 25
-o ./
-j result.jsondocker
The docker conatiner is
cgrlab/cgpbox_dev:develop
App ports
Inputs
inputs = list(
input(id = "bam", label = "bam", description = "either normal wildtype or tumour mutatantbam", type = "File", prefix = "-b", secondaryFiles = list(".bai")),
input(id = "ascat_adjusted_copynumber_csv", label = "ascat_adjusted_copynumber_csv", description = "ascat adjusted for caveman copynumber csv", type = "File", valueFrom = list("{if ($job.inputs.bam.metadata.sample_type == \"Tumor\"){return $self.path;}}"), prefix = "-a")
)arguments
arguments = CCBList(CommandLineBinding(position = 99, valueFrom = list("\"&& ls -lR\"")))Outputs
outputs = list(
output(id = "std_out", label = "std_out",
description = "standard output", type = "File...",
metadata = list(org = "cgp"),
glob = Expression(engine = "#cwl-js-engine",
script = "\"*.{out}\"")),
output(id = "verify_bam_result", label = "verify_bam_results",
description = "verify bam results", type = "File",
inheritMetadataFrom = "#bam", metadata = list(org = "cgp"),
glob = Expression(engine = "#cwl-js-engine",
script = "\"verify_bam/verify_bam_result.json\""))
)Define Tool Object and Push to Platform
Tool object
need to add some logic for -a adjusted copy number because it is only needed if it is the tumour sample.
-a /datastore/output/${NAME_MT}_vs_${NAME_WT}/ascat/${NAME_MT}.copynumber.caveman.csv
arguments = CCBList(CommandLineBinding(position = 2, prefix = "-a", valueFrom = list("\"if $self.metadata.sample_type = \"Tumor\" then \""))),
{if ($job.inputs.bam.metadata.sample_type == "Tumor") {command_line_arg = "ab.copynumber.caveman.csv";} else {command_line_arg = "";} return command_line_arg}
{if ($job.inputs.bam.metadata.sampleType == "Tumor") {command_line_arg = "ab.copynumber.caveman.csv";} else {command_line_arg = "";} return command_line_arg}
tool <- Tool(
id = "verify_bam",
label = "verify_bam",
hints = requirements(docker(pull = "cgrlab/cgpbox_dev:develop"), cpu(1), mem(1000)),
baseCommand = "verifyBamHomChk.pl -d 25 -o verify_bam -j verify_bam/verify_bam_result.json",
stdout = "std.out",
inputs = inputs, arguments = arguments, outputs = outputs)Push app to sbg platform
project$app_add("verify_bams", tool)4. ASCAT
Somatic copy number analysis using paired end wholegenome sequencing
https://github.com/cancerit/ascatNgs
base command
ascat.pl
-t [tumour bam]
-n [normal wildtype bam]
-r [genome reference]
-c [cpu number]
-rs 'HUMAN'
-ra GRCh37
-pr WGS
-pl ILLUMINA
-sg ascat_SnpGcCorrections.tsv
-q 20
-g L
-o ./ascat_outputPrep ASCAT for caveman
&& perl -ne '@F=(split q{,}, $_)[1,2,3,4]; $F[1]-1; print join("\t",@F)."\n";' < copynumber.caveman.csv > ./normal_cn.bed && perl -ne '@F=(split q{,}, $_)[1,2,3,6]; $F[1]-1; print join("\t",@F)."\n";' < copynumber.caveman.csv > ./tumour_cn.bed
docker
The docker conatiner is
cgrlab/cgpbox_dev:develop
App ports
Inputs
inputs = list(
input(id = "tumour_bam", label = "tumour_bam", description = "mutant tumour bam", type = "File", secondaryFiles = list(".bai"), prefix = "-t"),
input(id = "normal_bam", label = "normal_bam", description = "normal wildtype bam", type = "File", secondaryFiles = list(".bai"), prefix = "-n"),
input(id = "reference", label = "reference", description = "genome reference", type = "File", secondaryFiles = list(".fai"), prefix = "-r"),
input(id = "cgpbox_reference_files", label = "cgpbox_reference_files", description = "extracted reference files", type = "File...", stageInput = "link")
)arguments
arguments = CCBList(CommandLineBinding(position = 99, valueFrom = list("\"&& ls -lR\"")))Outputs
outputs = list(
output(id = "std_out", label = "std_out",
description = "standard output", type = "File...",
metadata = list(org = "cgp"),
glob = "\"std.out\""),
output(id = "ascat_statistics", label = "ascat_statistics",
description = "ascat_statistics", type = "File...",
inheritMetadataFrom = "#normal_bam", metadata = list(org = "cgp"),
glob = Expression(engine = "#cwl-js-engine",
script = "\"ascat_output/*.samplestatistics.txt\"")),
output(id = "copynumber_caveman", label = "copynumber_caveman",
description = "copy_number_caveman", type = "File...",
inheritMetadataFrom = "#normal_bam", metadata = list(org = "cgp"),
glob = Expression(engine = "#cwl-js-engine",
script = "\"ascat_output/*copynumber.caveman.csv\""))
)Add on scripts
prep_for_caveman_sh = fileDef(name = "prep_for_caveman.sh", content = read_file("prep_for_caveman.sh"))Define Tool Object and Push to Platform
Tool object
tool <- Tool(
id = "ASCAT",
label = "ASCAT",
hints = requirements(docker(pull = "cgrlab/cgpbox_dev:develop"), cpu(1), mem(1000)),
requirements = requirements(prep_for_caveman_sh),
baseCommand = "CPU=`grep -c ^processor /proc/cpuinfo` && ascat.pl -c $CPU -rs Human -ra NCBI37 -pr WGS -pl ILLUMINA -sg ascat_SnpGcCorrections.tsv -q 20 -g L -o ascat_output",
stdout = "std.out",
inputs = inputs,
arguments = arguments,
outputs = outputs)Push app to sbg platform
project$app_add("ASCAT", tool)5. CGP Pindel
Cancer Genome Project Insertion/Deletion detection pipeline based around Pindel
https://github.com/cancerit/cgpPindel
base command
pindel.pl
-t [tumour mutant bam]
-n [normal wildtype bam]
-r [genome reference]
-c $CPU
-sp HUMAN
-as GRCh37
-st WGS
-e NC_007605,hs37d5,GL%
-s pindel_simpleRepeats.bed.gz
-f pindel_genomicRules.lst
-g pindel_human.GRCh37.indelCoding.bed.gz
-u pindel_pindel_np.gff3.gz
-sf pindel_softRules.lst
-b brass-ucscHiDepth_0.01_mrg1000_no_exon_coreChrs.bed.gz
-o ./pindel_outputdocker
The docker conatiner is
cgrlab/cgpbox_dev:develop
App ports
Inputs
inputs = list(
input(id = "tumour_bam", label = "tumour_bam", description = "tumour bam", type = "File", secondaryFiles = list(".bai"), prefix = "-t"),
input(id = "normal_bam", label = "normal_bam", description = "wildtype bam", type = "File", secondaryFiles = list(".bai"), prefix = "-n"),
input(id = "reference", label = "reference", description = "reference", type = "File", secondaryFiles = list(".fai"), prefix = "-r"),
input(id = "cgpbox_reference_files", label = "cgpbox_reference_files", description = "extracted reference files", type = "File...", stageInput = "link")
)arguments
arguments = CCBList(CommandLineBinding(position = 99, valueFrom = list("\"&& ls -lR\"")))Outputs
outputs = list(
output(id = "std_out", label = "std_out",
description = "standard output", type = "File...",
metadata = list(org = "cgp"),
glob = "\"std.out\""),
output(id = "pindel_germline_bed", label = "pindel_germline_bed",
description = "pindel_germline_bed output", type = "File",
inheritMetadataFrom = "#tumour_bam", metadata = list(org = "cgp"),
glob = Expression(engine = "#cwl-js-engine",
script = "\"pindel_output/*.germline.bed\"")),
output(id = "pindel_flagged_vcf", label = "pindel_flagged_vcf",
description = "pindel_flagged_vcf", type = "File",
inheritMetadataFrom = "#tumour_bam", metadata = list(org = "cgp"),
glob = Expression(engine = "#cwl-js-engine",
script = "\"pindel_output/*.flagged.vcf.gz\""), secondaryFiles = list(".tbi"))
)Define Tool Object and Push to Platform
Tool object
tool <- Tool(
id = "pindel",
label = "pindel",
hints = requirements(docker(pull = "cgrlab/cgpbox_dev:develop"), cpu(1), mem(1000)),
baseCommand = "CPU=`grep -c ^processor /proc/cpuinfo` && pindel.pl -c $CPU -sp HUMAN -as GRCh37 -st WGS -e NC_007605,hs37d5,GL% -s pindel_simpleRepeats.bed.gz -f pindel_genomicRules.lst -g pindel_human.GRCh37.indelCoding.bed.gz -u pindel_pindel_np.gff3.gz -sf pindel_softRules.lst -b brass-ucscHiDepth_0.01_mrg1000_no_exon_coreChrs.bed.gz -o pindel_output",
stdout = "std.out",
inputs = inputs,
arguments = arguments,
outputs = outputs)Push app to sbg platform
project$app_add("pindel", tool)6. CaVEMan
CaVEMan actually needs the reference index and not the reference (?). But we can attach the reference index as a secondary file to the reference and find the path that way. Need to ask Erik about this.
It uses outputs from Pindel and ASCAT.
base command
caveman.pl
-tb [tumour mutant bam]
-nb [normal wildtype bam]
-r [genome reference index - genome.fa.fai]
-in [pindel .germline.bed]
-tc [tum.cn.bed - from ASCAT]
-nc [norm.cn.bed - from ASCAT]
-t $CPU
-s HUMAN
-sa GRCh37
-st WGS
-st genomic
-ig caveman_ucscHiDepth_0.01_merge1000_no_exon.tsv
-b ./
-u ./
-o caveman_outputdocker
The docker conatiner is
cgrlab/cgpbox_dev:develop
App ports
Inputs
inputs = list(
input(id = "tumour_bam", label = "tumour_bam", description = "mutant tumour bam", type = "File", secondaryFiles = list(".bai"), prefix = "-tb"),
input(id = "normal_bam", label = "normal_bam", description = "normal wildtype bam", type = "File", secondaryFiles = list(".bai"), prefix = "-nb"),
input(id = "genome_reference", label = "genome_reference", description = "genome_reference", type = "File", secondaryFiles = list(".fai"), valueFrom = list("$self.path+\".fai\""), prefix = "-r"),
input(id = "pindel_germline_bed", label = "pindel_germline_bed", description = "pindel_germline_bed", type = "File", prefix = "-in"),
input(id = "ascat_tumour_cn_bed", label = "ascat_tumour_cn_bed", description = "ascat_tumour_cn_bed", type = "File", prefix = "-tc"),
input(id = "ascat_normal_cn_bed", label = "ascat_normal_cn_bed", description = "ascat_normal_cn_bed", type = "File", prefix = "-nc"),
input(id = "cgpbox_reference_files", label = "cgpbox_reference_files", description = "extracted reference files", type = "File...", stageInput = "link")
)arguments
arguments = CCBList(CommandLineBinding(position = 99, valueFrom = list("\"&& ls -lR\"")))Outputs
outputs = list(
output(id = "std_out", label = "std_out",
description = "standard output", type = "File",
metadata = list(org = "cgp"),
glob = "\"std.out\""),
output(id = "caveman_snv_bed", label = "caveman_snv_bed",
description = "caveman_snv_bed", type = "File...",
inheritMetadataFrom = "#normal_bam", metadata = list(org = "cgp"),
glob = Expression(engine = "#cwl-js-engine",
script = "\"caveman_output/*bed\"")),
output(id = "caveman_all_vcf", label = "caveman_all_vcf",
description = "caveman_all_vcf", type = "File...",
inheritMetadataFrom = "#normal_bam", metadata = list(org = "cgp"),
glob = Expression(engine = "#cwl-js-engine",
script = "\"caveman_output/*vcf.gz\""), secondaryFiles = list(".tbi")),
output(id = "caveman_flagged_muts_vcf", label = "caveman_flagged_muts_vcf",
description = "caveman_flagged_muts_vcf", type = "File",
inheritMetadataFrom = "#normal_bam", metadata = list(org = "cgp"),
glob = Expression(engine = "#cwl-js-engine",
script ="\"caveman_output/*flagged.muts.vcf.gz\""), secondaryFiles = list(".tbi"))
)Define Tool Object and Push to Platform
Tool object
tool <- Tool(
id = "CaVEMan",
label = "CaVEMan",
hints = requirements(docker(pull = "cgrlab/cgpbox_dev:develop"), mem(1000)),
baseCommand = "CPU=`grep -c ^processor /proc/cpuinfo` && caveman.pl -t $CPU -s HUMAN -sa GRCh37 -st WGS -st genomic -ig caveman_ucscHiDepth_0.01_merge1000_no_exon.tsv -b ./ -u ./ -o caveman_output",
stdout = "std.out",
inputs = inputs,
arguments = arguments,
outputs = outputs)Push app to sbg platform
project$app_add("CaVEMan", tool)notes
needs these 3 inputs from other tools
-in /datastore/output/${NAME_MT}_vs_${NAME_WT}/pindel/${NAME_MT}_vs_${NAME_WT}.germline.bed \
-tc $TMP/tum.cn.bed \
-nc $TMP/norm.cn.bed \Prep ASCAT for caveman
echo -e "CaVEMan prep: `date`"
set -x
ASCAT_CN="/datastore/output/${NAME_MT}_vs_${NAME_WT}/ascat/$NAME_MT.copynumber.caveman.csv"
perl -ne '@F=(split q{,}, $_)[1,2,3,4]; $F[1]-1; print join("\t",@F)."\n";' < $ASCAT_CN > $TMP/norm.cn.bed
perl -ne '@F=(split q{,}, $_)[1,2,3,6]; $F[1]-1; print join("\t",@F)."\n";' < $ASCAT_CN > $TMP/tum.cn.bed
set +x
7. Brass
Breakpoints via assembly - Identifies breaks and attempts to assemble rearrangements.
https://github.com/cancerit/BRASS
base command
brass.pl
-t [tumour bam]
-n [normal wildtype bam]
-g [genome reference]
-ss [ascat *.samplestatistics.txt array]
-j 4
-k 4
-c $CPU
-s HUMAN
-as GRCh37
-pr WGS
-pl ILLUMINA
-d brass-ucscHiDepth_0.01_mrg1000_no_exon_coreChrs.bed.gz
-f brass-brass_np.groups.gz
-g_cache vagrent_Homo_sapiens.GRCh37.75.vagrent.cache.gz
-vi brass-viral.1.1.genomic.fa
-mi brass-all_ncbi_bacteria.20150703
-b brass-hs37d5_500bp_windows.gc.bed.gz
-ct brass-Human.GRCh37.CentTelo.tsv
-o ./brass_outputdocker
The docker conatiner is
cgrlab/cgpbox_dev:develop
App ports
Inputs
Need to add more inputs here.
inputs = list(
input(id = "tumour_bam", label = "tumour_bam", description = "tumour bam", type = "File", stageInput = "link", secondaryFiles = list(".bai"), valueFrom = list("{return $self.name}"), prefix = "-t"),
input(id = "normal_bam", label = "normal_bam", description = "wildtype bam", type = "File", stageInput = "link", secondaryFiles = list(".bai"), valueFrom = list("{return $self.name}"), prefix = "-n"),
input(id = "genome_reference", label = "genome_reference", description = "genome_reference", type = "File", secondaryFiles = list(".fai"), prefix = "-g"),
input(id = "bam_stats", label = "bam_stats", description = "bam stats", type = "File...", stageInput = "link"),
input(id = "ascat_sample_statistics", label = "ascat_sample_statistics", description = "ascat_sample_statistics", type = "File...", prefix = "-ss"),
input(id = "cgpbox_reference_files", label = "cgpbox_reference_files", description = "extracted reference files", type = "File...", stageInput = "link")
)arguments
arguments = CCBList(CommandLineBinding(position = 99, valueFrom = list("\"&& ls -lR\"")))Outputs
outputs = list(
output(id = "std_out", label = "std_out",
description = "standard output", type = "File...",
metadata = list(org = "cgp"),
glob = "\"std.out\""),
output(id = "brass_sv_bedpe", label = "brass_sv_bedpe",
description = "brass_sv_bedpe", type = "File",
inheritMetadataFrom = "#normal_bam", metadata = list(org = "cgp"),
glob = Expression(engine = "#cwl-js-engine",
script = "\"brass_output/*bedpe.gz\""), secondaryFiles = list(".tbi")),
output(id = "brass_vcf", label = "brass_vcf",
description = "brass_vcf", type = "File...",
inheritMetadataFrom = "#normal_bam", metadata = list(org = "cgp"),
glob = Expression(engine = "#cwl-js-engine",
script = "\"brass_output/*vcf*\""))
)Define Tool Object and Push to Platform
Tool object
tool <- Tool(
id = "BRASS",
label = "BRASS",
hints = requirements(docker(pull = "cgrlab/cgpbox_dev:develop"), cpu(1), mem(1000)),
baseCommand = "CPU=`grep -c ^processor /proc/cpuinfo` && brass.pl -j 4 -k 4 -c $CPU -s HUMAN -as GRCh37 -pr WGS -pl ILLUMINA -d brass-ucscHiDepth_0.01_mrg1000_no_exon_coreChrs.bed.gz -f brass-brass_np.groups.gz -g_cache vagrent_Homo_sapiens.GRCh37.75.vagrent.cache.gz -vi brass-viral.1.1.genomic.fa -mi brass-all_ncbi_bacteria.20150703 -b brass-hs37d5_500bp_windows.gc.bed.gz -ct brass-Human.GRCh37.CentTelo.tsv -o ./brass_output",
stdout = "std.out", inputs = inputs, arguments = arguments, outputs = outputs)Push app to sbg platform
project$app_add("BRASS", tool)8. CGP Annotate VCF
CGP Annotate
base command
AnnotateVcf.pl
-i [VCF input from pindel or caveman]
-c vagrent_Homo_sapiens.GRCh37.75.vagrent.cache.gz
-o annotated.muts.vcfdocker
The docker conatiner is
cgrlab/cgpbox_dev:develop
App ports
Inputs
inputs = list(
input(id = "vcf", label = "vcf", description = "vcf prep annotation", type = "File", secondaryFiles = list(".tbi"), prefix = "-i"),
input(id = "cgpbox_reference_files", label = "cgpbox_reference_files", description = "extracted reference files", type = "File...", stageInput = "link")
)arguments
arguments = arguments = CCBList(CommandLineBinding(position = 99, valueFrom = list("\"&& ls -lR\"")))Outputs
outputs = list(
output(id = "std_out", label = "std_out",
description = "standard output", type = "File...",
metadata = list(org = "cgp"),
glob = "\"std.out\""),
output(id = "annotated_vcf", label = "annotated_vcf",
description = "annotated vcf", type = "File",
inheritMetadataFrom = "#vcf", metadata = list(org = "cgp"),
glob = "\"*vcf*\"")
)Define Tool Object and Push to Platform
Tool object
tool <- Tool(
id = "cgp_annotate_vcf",
label = "CGP Annotate VCFs",
hints = requirements(docker(pull = "cgrlab/cgpbox_dev:develop"), cpu(1), mem(1000)),
baseCommand = "AnnotateVcf.pl -c vagrent_Homo_sapiens.GRCh37.75.vagrent.cache.gz -o annotated.muts.vcf",
stdout = "std.out",
inputs = inputs,
arguments = arguments,
outputs = outputs)Make cwl file
write(tool$toJSON(pretty = TRUE), "cgp_annotate_vcf.json")Push app to sbg platform
project$app_add("cgp_annotate_vcf", tool)notes
# annotate pindel
AnnotateVcf.pl
-t
-c $REF_BASE/vagrent/vagrent.cache.gz
-i /datastore/output/${NAME_MT}_vs_${NAME_WT}/pindel/${NAME_MT}_vs_${NAME_WT}.flagged.vcf.gz
-o /datastore/output/${NAME_MT}_vs_${NAME_WT}/pindel/${NAME_MT}_vs_${NAME_WT}.annot.vcf
# annotate caveman
AnnotateVcf.pl
-t
-c $REF_BASE/vagrent/vagrent.cache.gz \
-i /datastore/output/${NAME_MT}_vs_${NAME_WT}/caveman/${NAME_MT}_vs_${NAME_WT}.flagged.muts.vcf.gz
-o /datastore/output/${NAME_MT}_vs_${NAME_WT}/c.annot.muts.vcf